¿Qué es la Piel de Mariposa?

La Piel de Mariposa o Epidermólisis bullosa es un término que engloba a un grupo clínico y genéticamente heterogéneo de enfermedades genéticas ampollosas de baja prevalencia, cuya principal característica es una extrema fragilidad de la piel y de las membranas mucosas. Este trastorno da lugar a la formación de ampollas y heridas ante mínimos traumatismos incluso de forma espontánea.1
La EB es una enfermedad multisistémica y crónica que impacta gravemente en la calidad de vida de la persona afectada y sus familiares.
¿Cuántas personas sufren Piel de Mariposa?
Se estima que, a nivel mundial, 500.000 personas tienen Epidermólisis Bullosa. En España, la prevalencia es de 1.000 personas (2,4 personas por 100.000 habitantes). Este grupo de enfermedades afecta a ambos sexos por igual y todas las etnias.2
Conoce el compromiso que tiene Urgo Medical con las personas que padecen de esta patología en este enlace.
¿Cómo se produce la Piel de Mariposa?
La piel consta de una serie de capas: epidermis, dermis e hipodermis. La epidermis es la capa más superficial de la piel y está formada por células estrechamente unidas gracias a una serie de proteínas. Si hiciéramos un símil, podríamos comparar la epidermis con una “pared de ladrillo”, en el que cada célula es un “ladrillo” y las proteínas el “cemento” que une toda la estructura.1
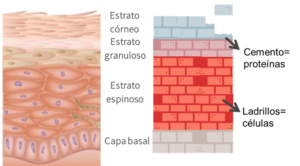
La Epidermólisis Bullosa se produce por mutaciones en los genes que codifican estas proteínas. Como resultado, perdemos el “cemento” que une las células produciendo que se desprendan ante mínimos traumatismos.
¿Cuáles son los tipos de Epidermólisis Bullosa?
Existe diferentes tipos de Epidermólisis Bullosa en función de la capa de la piel que se vea afectada. La más frecuente es la EB Simple que se produce por alteración de las proteínas situadas en la epidermis. La EB juntural afecta a aquellas proteínas situadas en la lámina basal (capa que marca el límite entre la dermis y la epidermis), mientras que en la EB Distrófica ocurre por lesiones a nivel de la dermis.3
Por último, existe un fenotipo que es la EB Kindler que se considera una enfermedad independiente ya que la formación de ampollas no tiene lugar en una capa concreta de la piel sino que se puede producir indistintamente en cualquiera de las capas.4
¿Cuál es el tratamiento de la Epidermólisis Bullosa?
El objetivo terapéutico en los pacientes con epidermólisis bullosa (EB) está centrado en prevenir la incidencia de heridas crónicas y conseguir la mejoría en la cicatrización de las mismas, acelerando los procesos que ocurren a nivel celular.
Una de las tecnologías que ha demostrado una buena aceptabilidad, alta tolerancia y eficacia en los pacientes con EB es la Tecnología Lipidocoloide (TLC). La eficacia de la TLC se ha demostrado a través de un ensayo clínico de 20 pacientes (11 adultos, 9 niños) con EB Simple o Distrófica durante 4 semanas. En el estudio se registraron todos los cambios de apósito, los parámetros de la herida, el dolor y el efecto del apósito sobre la calidad de vida de los pacientes. 5
Los principales resultados del estudio fueron5:
- 19 de 20 heridas se curaron en 8,7 ± 8,5 días.
- 11 pacientes (55%) consideraron que su calidad de vida había mejorado tras el uso del apósito.
- La tecnología TLC se consideró indolora y «muy fácil» o «fácil» de retirar en la mayoría de los cambios de apósito.
- 19 de 20 pacientes afirmaron que utilizarían el apósito del estudio para tratar sus lesiones en el futuro.
Este estudio confirmó la muy buena aceptabilidad y eficacia de la tecnología TLC en el tratamiento de las lesiones cutáneas en pacientes con EB.5
A continuación, te presentamos un ejemplo de la evolución de una herida tratado con la con tecnología TLC:

Afectado de epidermólisis bullosa tratado con un apósito con tecnología TLC. 5,6
Si quieres obtener más información sobre la Epidermólisis Bullosa sobre cómo tratar con pacientes que tiene esta patología o algunos consejos ve a nuestra sección de VIVIR CON UNA HERIDA.
Referencias
- Fine J. Inherited epidermolysis bullosa. Orphanet journal of rare diseases 2010;5(1):1;
- Mariposa, P.de (2020) Introducción a la epidermólisis bullosa, Asociación DEBRA-PIEL DE MARIPOSA. Available at: https://www.pieldemariposa.es/introduccion-a-la-epidermolisis-bullosa/ (Accessed: October 31, 2022).
- Mariposa, P.de (2020) ¿Qué tipos de epidermólisis bullosa existen?, Asociación DEBRA-PIEL DE MARIPOSA. Available at: https://www.pieldemariposa.es/que-tipos-de-epidermolisis-bullosa-existen/ (Accessed: October 31, 2022).
- Síndrome de Kindler (KS) (no date) EB House Austria. Available at: https://www.eb-haus.org/es/manual-de-eb/la-enfermedad/sindrome-de-kindler-ks/ (Accessed: October 31, 2022).
- Blanchet-Bardon C, er al Using Urgotul dressing for the management of epidermolysis bullosa skin lesions. Journal of Wound Care. 2005;14(10):490-496.
- Bourdon-Lanoy E., Corset I. et al. Observation of the new UrgoTul Flex dressing in the local treatment of hereditary Epidermolysis Bullosa lesions. Poster, CPC 2010, Paris.